


- 地址:
- 贵州省贵阳市国家高新技术产业开发区长岭南路178号茅台国际商务中心一期第AB幢(B)1单元9层19号房
- 邮箱:
- yg8174@qq.com
- QQ:
- 259633603
- 传真:
- 0851-84851236
- 手机:
- 18606505966
全球首例!6个月婴儿接受定制基米乐(MILE)-官方首页因编辑治疗 罕见病治疗新突破。美国费城儿童医院和宾夕法尼亚大学医学院的研究团队最近取得了一项具有里程碑意义的医学突破——人类首次实现为单个病人定制基因编辑疗法,并成功治疗了一名患有罕见致命遗传疾病的婴儿。这项研究于2025年5月15日发表在《新英格兰医学杂志》上,详细介绍了这款定制的体内碱基编辑疗法的开发和治疗过程。

这一成功案例可能为基因编辑技术开辟一条应用于治疗那些目前尚无医疗手段可用的罕见病患者的新途径。婴儿KJ Muldoon在出生几天后被诊断出患有严重的氨甲酰磷酸合成酶-1(CPS1)缺乏症。这是一种罕见且严重的隐性遗传疾病,在新生儿中的发病率为130万分之一,是最严重的尿素循环障碍疾病之一。人体在分解蛋白质时会产生氨,正常情况下会将氨转化为尿素并排出体外。CPS1功能缺失会导致氨迅速在血液中积聚,从而导致器官损伤,尤其是大脑和肝脏,若没有得到及时治疗,可能会迅速进展为昏迷甚至死亡。据统计,CPS1缺乏症患者在婴儿早期的死亡率高达50%。

CPS1基因很大,超过了腺相关病毒(AAV)载体的递送极限,此外,婴儿的肝细胞更替速度较快,导致递送到肝脏的载体会被快速稀释。因此,该疾病难以通过传统的递送正确版本的CPS1基因进行治疗。目前,该疾病的治疗手段有限,包括米乐(MILE)-官方首页透析、氨清除剂、限制蛋白质摄入以及晚期肝移植,但这些方法难以阻止氨对大脑神经系统的损伤。
不久前,美国FDA批准了基于CRISPR-Cas9的基因编辑疗法Casgevy上市,用于治疗两种相对常见的遗传疾病——镰状细胞病(SCD)和β-地中海贫血(TDT)。碱基编辑是刘如谦教授基于CRISPR开发的新一代基因编辑技术,其不依赖DNA双链断裂,能够精准修复人类基因组中的致病突变。
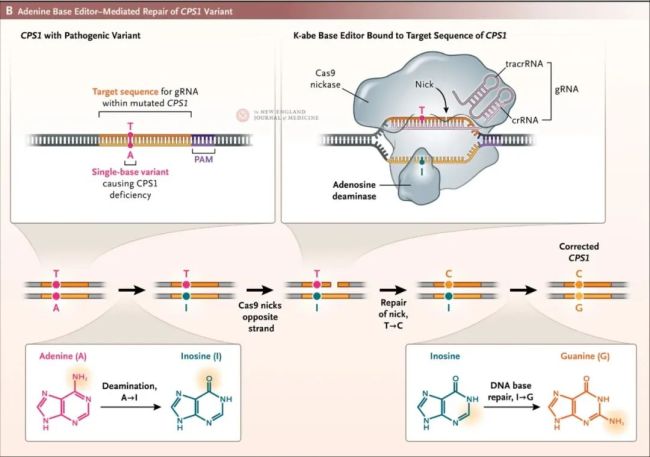
研究团队通过对KJ基因组的快速靶向分析,发现他来自父亲和母亲的CPS1基因分别有一个单碱基突变(Q335X和E714X),这导致了CPS1的两个等位基因在编码蛋白时提前终止。这种基因突变情况非常适合使用碱基编辑技术进行修复。研究团队联合学术界和工业界,启动了定制碱基编辑疗法的开发工作,从细胞和小鼠模型构建开始,进行了体外和体内动物实验,并在猴子中进行了安全性和有效性验证,最终开发出了这款定制的脂质纳米颗粒(LNP)递送的碱基编辑疗法——kayjayguran abengcemeran(k-abe),利用腺嘌呤碱基编辑器(ABE)来修复患者CPS1基因中的单碱基突变Q335X。
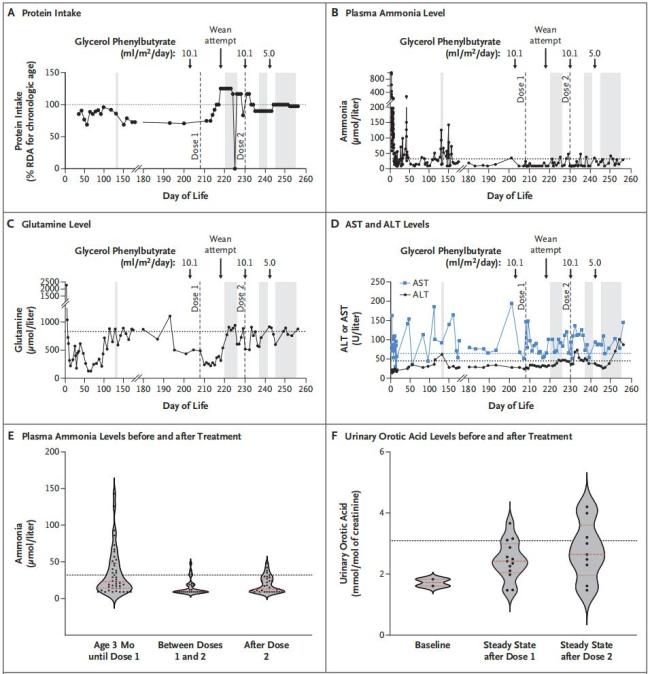
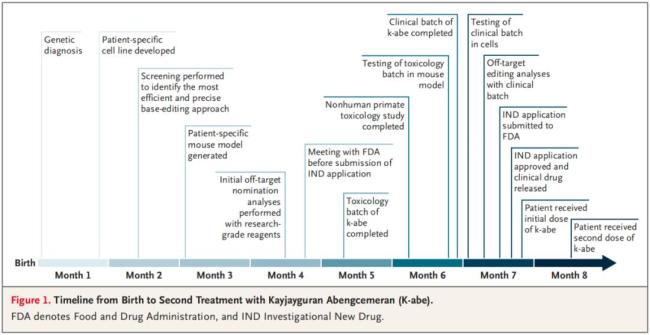
整个开发、验证、生产以及监管审批过程仅花费六个月时间。在此期间,KJ在医院度过,每天服用氨清除剂,并严格遵循特殊的极低蛋白饮食。2025年2月底,在得到监管审批后,KJ接受了这种实验性碱基编辑疗法的首次输注,3月和4月又接受了后续剂量的治疗。截至2025年4月,KJ已接受了三次治疗,未出现严重副作用。治疗后,他能够耐受更多的蛋白质摄入,所需的氨清除剂用量减半,还能够从某些常见的儿童疾病(例如鼻病毒感染)中恢复,而体内氨水平不会升高。他的体重也开始稳步提升。研究团队将继续对其进行长期随访,以全面评估该疗法的益处。

目前,即将十个月大的KJ正在茁壮成长。每达成一个小小的里程碑,对他的父母而言都像是一个小小的奇迹。就在上周,他的母亲走进KJ的病房,发现他正独自坐在婴儿床里,她激动地表示:我们从没想过这会发生。
